
 Rīga +1°C
Rīga +1°C 


Novērtē rakstu
-
 0
0
-
 0
0
-
 0
0
-
 0
0
-
 0
0
-
 0
0
-
 0
0
-

Piemēram, Fabrī slimības pacientiem bieži vērojama arī karstuma nepanesība, daļai ar laiku parādās izmaiņas ādā – nelielas sarkanvioletas piepaceltas papulas jeb angiokeratomas. Ja laikus ir noteikta pareiza diagnoze, arī ģenētisku slimību iespējams sekmīgi ārstēt, lai ar to varētu sadzīvot.
Klīniskā ģenētiķe Zita Krūmiņa (VCA poliklīnikā Elite) norāda, ka ir vairāk nekā piecdesmit iedzimtas vielmaiņas slimības, kas pieder pie lizosomālajām uzkrāšanās slimībām. Šoreiz gan tikai par trim – Gošē slimību, Fabrī slimību un Pompes slimību. Tām visām slimības manifestācija novērojama galvenokārt pieaugušā vecumā.
Kas īsti notiek organismā?
“Katrā šūnā ir daudz organoīdu, piemēram, ir mitohondriji, kas dod enerģiju, šūnas kodols, kurā atrodas šūnas ģenētiskais materiāls (DNS), plazmatiskā membrāna, kas šūnu norobežo no āršūnas vides un citi. Un ir arī lizosomas ar dažādām funkcijām. Viena no tām – uzturvielu (makromolekulu) noārdīšana. Ja lizosomas enzīmu trūkuma dēļ nespēj uzturvielas sašķelt, šie starpprodukti uzkrājas kā neiznesti atkritumu maisi, kas ar laiku piepilda visu šūnu līdz tā aiziet bojā, izraisot lēni progresējošas patoloģiskas izmaiņas dažādos orgānos.
Lai gan trīs nosauktās slimības pieder pie vienas slimību grupas, tās ir atšķirīgas, un pat vienas slimības izpausmes dažādiem cilvēkiem var atšķirties,” skaidro Zita Krūmiņa.
Gošē slimība
“Gošē slimības gadījumā daudziem pacientiem novēro anēmiju, tomēr ne visiem – kādam ir zemāks hemoglobīns, taču biežāk novērojams zemāks trombocītu skaits. Kā slimības simptoms var būt asinsizplūdumi ādā, arī biežākas deguna asiņošanas epizodes. Biežākā izpausme (aptuveni 80% gadījumu) ir neskaidras izcelsmes palielināts liesas izmērs. Vairāk nekā pusei cilvēku ar šo diagnozi ir arī palielinātas aknas, reizēm vērojamas izmaiņas aknu funkcijās. Trešā izplatītākā pazīme ir izmaiņas kaulu struktūrā, reizēm arī stipras sāpes (biežāk – gūžas locītavā), kas var atgādināt akūtu osteomelītu,” stāsta ārste.
Vidējais vecums, kad parādās kāds no šiem simptomiem, ir 25–28 gadi. Tiesa, ir gadījumi, kad Gošē slimība izpaužas jau divu, trīs gadu vecumā ar tādu pazīmi, kā liels vēders. Izmeklējot konstatē, ka ir palielināta liesa un aknas, zems hemoglobīns un trombocītu skaits, pastiprināta zilumu veidošanās ādā.
Zita Krūmiņa norāda, ka reizēm pirmie Gošē slimības simptomi sākas tikai pēc četrdesmit gadu vecuma – parādās ostopēnija, bieži kaulu lūzumi, izmaiņas kaulu struktūrā.
Viena no šīs slimības īpatnībām ir tā, ka aškenazi ebreju (lielākā ebreju tautas daļa Centrālajā un Austrumeiropā) vidū tā ir sastopama salīdzinoši biežāk un to pat nevar uzskatīt par retu – viens gadījums uz 800 iedzīvotāju. Pārējās populācijās sastopams vidēji viens gadījums uz 40 000. Jāņem vērā, ka Eiropā par retām uzskata slimības, kuru izplatība ir retāka par vienu gadījumu uz 2000 iedzīvotāju.
Kā diagnosticē Gošē slimību?
“Diagnozes apstiprināšana ir vienkārša – aizdomu gadījumā ģimenes ārstam vai speciālistam ir tiesības nosūtīt pacientu uz Bērnu klīniskās universitātes slimnīcas Reto slimību koordinācijas centru, kur, ar pacienta piekrišanu, paņem asins pilienu no pirksta un nosūta uz laboratoriju ārzemēs, lai pārbaudītu enzīma aktivitāti. Analīze ir bez maksas, pieaugušajam ir jāsamaksā vien nepilnus divus eiro par asins paņemšanu.
Ja atklājas, ka ir samazināts enzīma daudzums, izmantojot to pašu asins materiālu, laboratorija ārzemēs arī veic molekulāro diagnostiku, atklājot slimību izraisošās mutācijas gēnā, kas apstiprina diagnozi. Slimības gadījumā pacientam ir jāmanto divas mutācijas noteiktajā gēnā. Viena mutācija var būt mantota no mātes, otra no tēva, kuri paši ir veseli, jo katram ir tikai viena patogēnā mutācija.”
Terapija, lai ārstētu Gošē slimību
Pirmreizēji apstiprinātus Gošē slimības pacientus (gan bērnus, gan pieaugušos) konsultē Bērnu klīniskās universitātes slimnīcas Reto slimību centrā, kur tiek lemts par papildus izmeklējumu nepieciešamību pacientam vai citiem ģimenes locekļiem. Viens no galveniem jautājumiem – vai ir indikācijas uzsākt specifisku enzīma aizvietojošu terapiju. To lemj ārstu konsīlijs, izvērtējot slimības gaitu, klīniskos un laboratoriskos datus.
Gošē slimībai ir trīs tipi. Aptuveni 92% gadījumu ir sastopams pirmais tips. Šī tipa ārstēšanai ir efektīva enzīma aizvietojošā terapija. “Enzīmu terapijas efektivitāte ir ļoti laba – pat pusgada laikā uzlabojas hematoloģiskie rādītāji, pakāpeniski mazinās aknu, liesas izmēri, ilgāks laika posms paiet kaulu struktūras labošanai. Tātad – patoloģiskie simptomi ir atgriezeniski. Veiksmīgas terapijas rezultātā var turpināt pilnvērtīgu, aktīvu dzīvi. Ja šo slimību konstatē jaunam cilvēkam un uzsāk enzīma aizvietojošo terapiju, tā neatstāj negatīvu iespaidu, piemēram, uz reproduktīvo veselību. Tiesa, jāņem vērā, ka pēcnācējs mantos vienu Gošē slimības izraisošo gēna mutāciju. Taču ir ļoti maza varbūtība, ka šis cilvēks izveidos attiecības ar partneri, kuram arī ir šī gēna mutācija. Ja tomēr sastopas divi cilvēki ar Gošē slimību, tad visiem viņu bērniem būs Gošē slimība.”
Fabrī slimība
Fabrī slimība ir lēni progresējoša multisistēmiska slimība, kad saistībā ar enzīma trūkumu šūnās uzkrājas glikosfingolipīdi. Visbiežāk to novēro nieru epitēlijšūnās, miokarda šūnās, asinsvadu endotēlijā, centrālās nervu sistēmas šūnās, ādas šūnās, acs radzenē.
VCA poliklīnikas Elite ģenētiķe skaidro, ka Fabrī slimība tiek pārmantota ar X hromosomu un slimot vajadzētu galvenokārt vīriešiem, jo viņiem ir tikai viena X hromosoma, bet sievietēm divas. “Par šo slimību joprojām ir daudz neskaidrību, jo aptuveni divām trešdaļām sieviešu, kuru vienā X hromosomā ir slimības gēns, arī ir slimības izpausmes, tikai klīniskie simptomi parasti sākas vēlāk un slimības norise biežāk ir bez smagām komplikācijām.
Fabrī slimību mēdz dēvēt par hameleonu ar daudz un dažādam nokrāsām: kādam pacientam ir vairāk izmaiņu, kas saistīti ar centrālo nervu sistēmu, piemēram, agrīns insults, citam ir sirds problēmas (biežāk – kreisā kambara hipertrofija), var būt arī sirds ritma traucējumi.
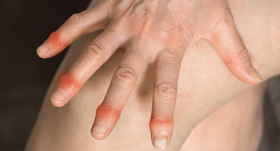
Slimība bieži vien sākas kā dedzinošas sāpes roku un kāju pirkstos (akroparestēzijas), kas ilgst stundas vai dienas un ko mēdz provocēt stress vai fiziska slodze. Bieži pacientiem vērojama karstuma nepanesība, daļai ar laiku parādās izmaiņas ādā – nelielas sarkanvioletas piepaceltas papulas jeb angiokeratomas, kas neniez, taču rada kosmētisku defektu. Biežāk tās vērojamas ap nabu, uz augšstilbiem, muguras, plaukstām. Kāda no šīm pazīmēm var parādīties jau pusaudžu vecumā un pakāpeniski pastiprināties. Daļai pacientu var būt sūdzības par kuņģa zarnu trakta darbību: caureja, sāpes vēderā, bet daļai pacientu pieaugušo vecumā veidojas vājdzirdība, kas var būt vienpusēja un pārejoša. Daudziem Fabrī slimības pacientiem var novērot arī radzenes un lēcas apduļķošanos, taču parasti tā neizraisa nozīmīgus redzes bojājumus.”
Nopietnas veselības problēmas, kas var izraisīt priekšlaicīgu nāvi, var būt insults, miokarda un sirds vārstuļu problēmas, kā arī progresējoša nieru nepietiekamība. Nieru mazspēja ir viens no Fabrī slimības pacientu galvenajiem nāves cēloņiem.
Kā diagnosticē Fabrī slimību?
Slimību diagnosticē, veicot enzīma aktivitātes noteikšanu asinīs un molekulāro diagnostiku (līdzīgi kā Gošē slimības gadījumā). “Problēma ir saistīta ar to, ka klīnisko simptomu daudzveidības dēļ ne ģimenes ārsti, ne citi speciālisti bieži neatpazīst Fabrī slimību. Iesaistīti ir tik daudzi orgāni, ka dažreiz paiet pat divdesmit gadi, apmeklējot daudzus speciālistus un veicot dažādus izmeklējumus, līdz izdodas nonākt pie šīs diagnozes. Nereti vienīgais izteikti traucējošais simptoms ir dedzinošas sāpes roku un kāju pirkstos. Parasti tad cilvēks vairākkārt apmeklē neirologu, kurš mēģina mazināt simptomus ar dažādiem medikamentiem,” stāsta Zita Krūmiņa.
Terapija, lai ārstētu Fabrī slimību
Par indikācijām enzīma aizvietojošai terapijai, lemj ārstu konsīlijs, izvērtējot klīniskās izpausmes, izmeklējuma rezultātu par enzīma aktivitāti. Pārbauda arī, cik daudz nesašķelto starpproduktu ir uzkrājušies asinīs. “Reizēm slimība ir tik tālu attīstījusies, ka bojājums nierēs ir neatgriezenisks un enzīma aizvietojošā terapija vairs nevar dot pozitīvu rezultātu. Ir gadījumi, kad, pacientam veikta nieru transplantācija vai hemodialīze vēl pirms ir apstiprināta Fabrī slimības diagnoze.”
Pompes slimība
Pompes slimības gadījumā lizosomās uzkrājas nesašķeltais glikogēns. Sākotnēji bieži vien ir uzstādīta cita diagnoze, piemēram: plecu-iegurņa joslas muskuļu distrofija, Bekera muskuļu distrofija, polimiozīts (muskuļu iekaisums), un tikai vēlāk izdodas precizēt, ka pie vainas ir Pompes slimība.
Šai iedzimtajai vielmaiņas slimībai ir divi tipi. Viens ir klasiskais zīdaiņu tips, kas sākas pirmajos dzīves mēnešos un noris ar sirds paplašināšanos (kardiomiopātiju), izteiktu hipotoniju un agrīnu elpošanas un sirds nepietiekamību. Ja slimība netiek savlaicīgi diagnosticēta un uzsākta terapija, dzīves ilgums parasti ir viens vai pusotrs gads.
“Biežāk sastopams ir Pompes slimības vēlīnais tips. Vidējais vecums, kad parādās klīniskie simptomi, ir 20–30 gadi, taču tie var izpausties arī agrākā vai vēlākā vecumā. Pirmie klīniskie simptomi, kas noris ar pakāpenisku muskuļu vājumu, parādās ap divdesmit gadu vecumu. Jaunietis, kurš iepriekš bijis aktīvs, sportojis, pamana, ka tas kļūst grūtāk. Pakāpeniski (desmit, piecpadsmit gadu laikā) pazīmes pastiprinās, līdz ir tik izteikts muskuļu vājums, ka tikai ar lielu piepūli var uzkāpt pa kāpnēm, ir grūti piecelties no krēsla, nepieciešamas papildierīces, reizēm – ratiņķrēsls,” slimības gaitu apraksta ģenētiķe.
Progresējot muskuļu vājumam, nereti rodas arī elpošanas nepietiekamība (elpošanu nodrošina diafragmas muskuļi) un ir nepieciešama papildu skābekļa pievade.
Kā diagnosticē Pompes slimību?
Marķieris asinīs, ko svarīgi pārbaudīt, ir kreatīnfosfokināze. Paaugstināts kreatīnfosfokināzes līmenis var liecināt par vairākām slimībām, taču ir arī pirmais signāls par iespējamu Pompes slimību. Ja tā, tad jāveic enzīma tests un jānoskaidro, vai enzīma aktivitāte ir normāla vai samazināta.
Terapija, lai ārstētu Pompes slimību
Kad Pompes slimības diagnoze ir apstiprināta, ārstu konsīlijs nolemj, vai ir lietderīgi uzsākt enzīma aizvietojošo terapiju. Ja slimību diagnosticē laikus, uzsākot enzīma aizvietojošo terapiju, var panākt slimības neprogresēšanu un klīnisko simptomu mazināšanos.
Enzīma aizvietojošo terapiju var saņemt tikai stacionārā. Ja ārstu konsīlijs ir lēmis par šīs terapijas nepieciešamību, izmaksas sedz valsts no reto slimību fonda.
“Ziņa par ģenētisku slimību biedē, tomēr, ja jebkura no šīm trim slimībām tiek laikus diagnosticēta un uzsākta rūpīga pacienta stāvokļa uzraudzīšana un atbilstoša enzīma aizvietojošā terapija, cilvēks var dzīvot pilnvērtīgu dzīvi un neizjust ģenētisko slimību kā ierobežojošu faktoru,” norāda ģenētiķe Zita Krūmiņa.